PIGTを責任遺伝子とする発作性夜間ヘモグロビン尿症(PNH)の発症メカニズム解明(木下研がJCIに発表)
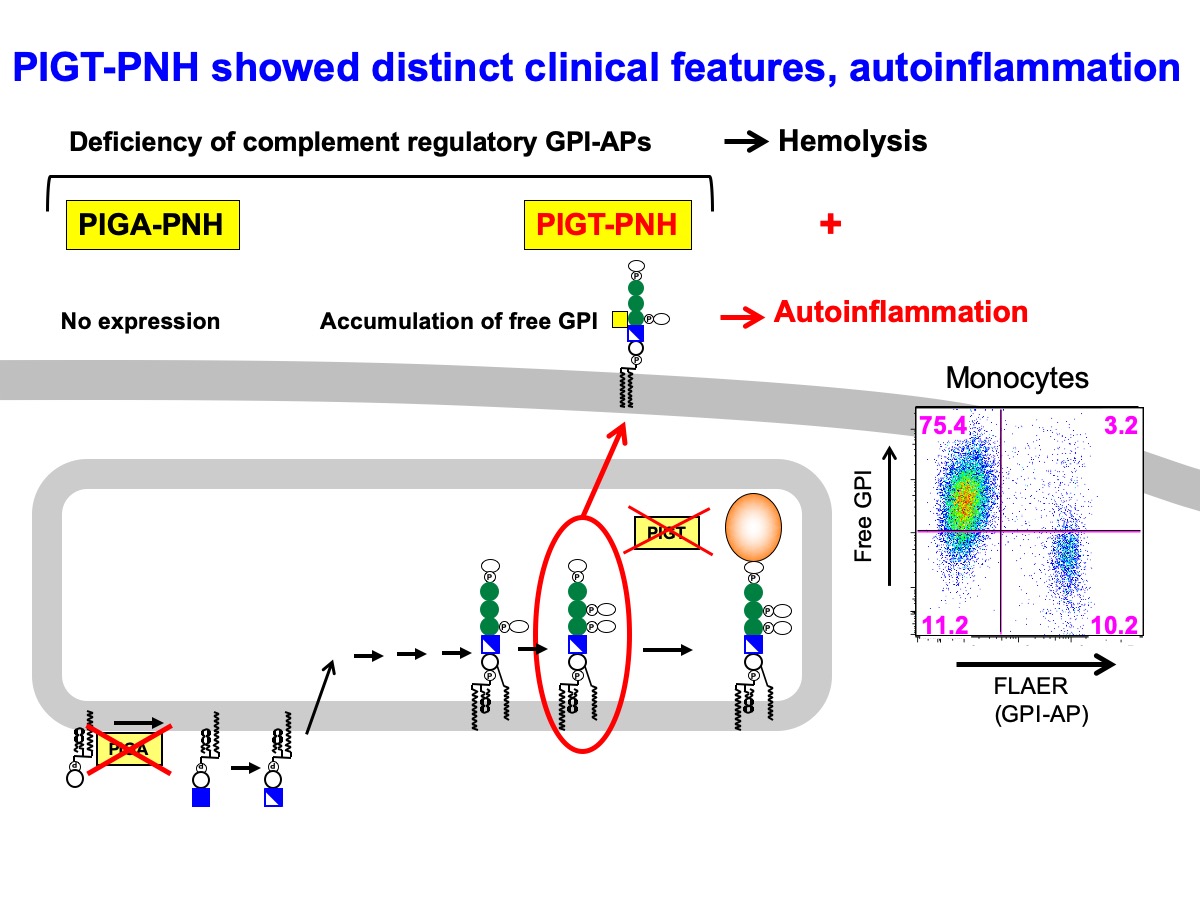
発作性夜間ヘモグロビン尿症(PNH)は造血幹細胞のX染色体上のPIGA遺伝子の突然変異により発症する血液疾患で溶血発作を主症状とする。GPI-AP生合成と修飾に関わる遺伝子は27個同定されているが、大部分のPNH患者の原因遺伝子はPIGAである。その理由はPIGAのみがX染色体上の遺伝子で男女とも機能的アレルは1本で1回の体細胞突然変異でGPI欠損細胞になるためと考えられる。今回、我々はPIGTを原因遺伝子とするPIGT-PNHの4症例を報告した。患者はいずれも遺伝的に一方のアレルに変異があるところに、造血幹細胞において体細胞突然変異による20番染色体のPIGT周辺領域の欠損が起こって発症したものであった。この染色体20qの領域は骨髄増殖性疾患で共通して欠損している領域を含んでおりこの部位にあるインンプリンティング遺伝子の欠損が異常クローン拡大に関与していると考えられた。国内症例ではPNHの診断前15年以上にわたって蕁麻疹、発熱、関節痛、腹痛、無菌性髄膜炎などの自己炎症症状を来していたが、PNH診断後抗補体薬であるエクリズマブの投与によりこれらの炎症も治まった。このようにPIGT-PNHはPIGA-PNHに見られない自己炎症症状が特徴的である。この症状の違いは、PIGA欠損ではGPI合成が最初のステップで止まるのに対し、PIGTは前駆タンパク質にGPIアンカーを付加する反応に関わるため、その欠損ではタンパク質に付かないGPIアンカー(フリー GPI) が蓄積し細胞表面に発現しており、これが補体活性化と共にインフラマソームの活性化をきたし炎症を誘導するためである。ヒトの単球細胞株であるTHP-1細胞のPIGT-KO細胞ではレクチン経路の活性化により補体の活性化がより増幅され、膜障害複合体(MAC)形成が促進されて大量のIL-1βが遊離されることがわかった。このようにPIGT-PNHは特徴的な臨床症状に加えて異常クローンの拡大メカニズムについてもPIGA-PNHと異なる新しいPNHの病型である。
本研究はThe Journal of Clinical Investigation誌に2019年8月に掲載されました。
Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation.
Höchsmann B, Murakami Y, Osato M, Knaus A, Kawamoto M, Inoue N, Hirata T, Murata S, Anliker M, Eggermann T, Jäger M, Floettmann R, Höellein A, Murase S, Ueda Y, Nishimura JI, Kanakura Y, Kohara N, Schrezenmeier H, Krawitz PM, Kinoshita T.