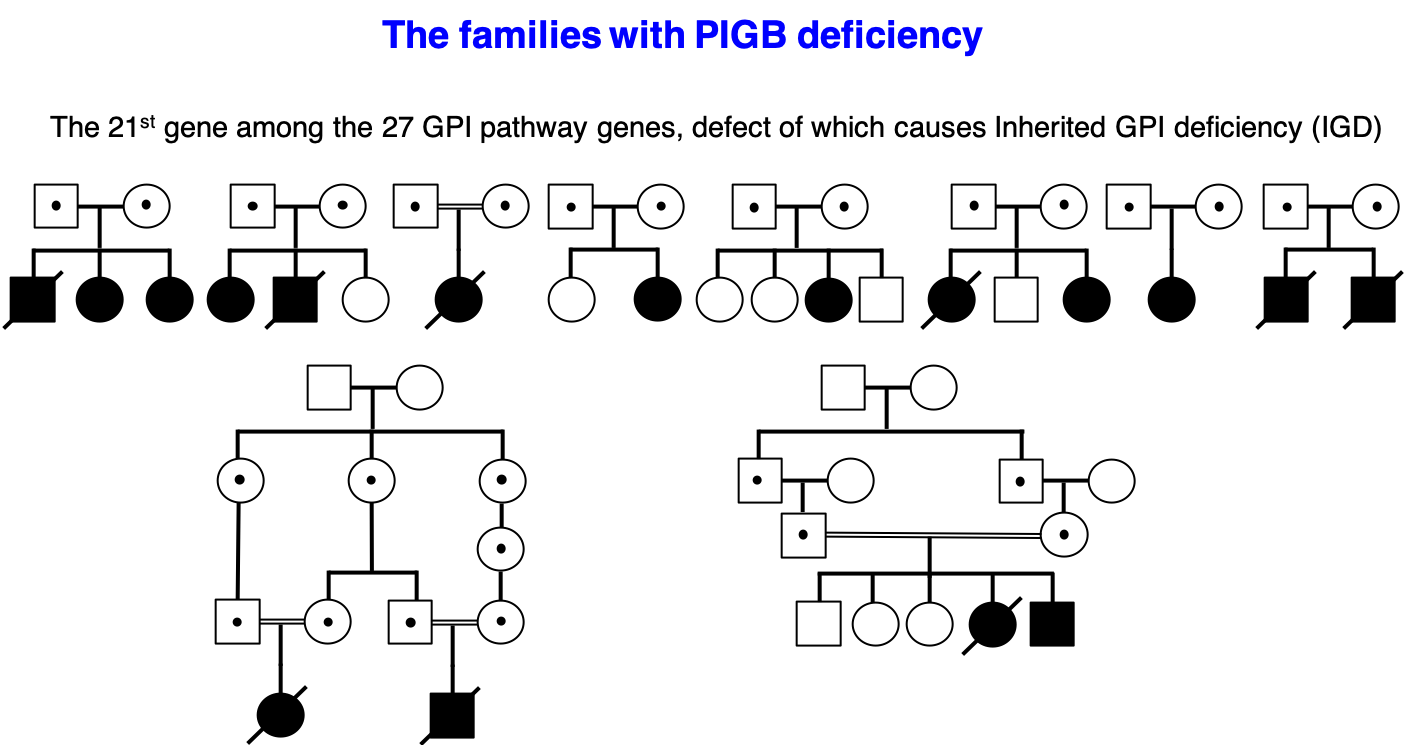
新しい先天性GPI欠損症「PIGB 欠損症」を発見(木下研がAJHG誌に発表)
種々のGPI アンカー型タンパク質(GPI-AP)は生体において様々な機能を担っているが特に形態形成や神経発達に重要な役割を持つ。GPI-APの生合成と修飾、輸送に係わる遺伝子は27個あり、これらの遺伝子の変異により,精神運動発達の遅れやてんかんを主症状とする先天性GPI欠損症(IGD)を来し、現在は国内では指定難病に認定されている。今までに20種の遺伝子異常によるIGDが報告されており、変異遺伝子の機能やその活性低下の度合いにより様々な症状を呈する。我々はドイツ・イギリス・カナダの研究者と共同研究をしており、現在までに多くのIGDを報告している。今回の報告はカナダのモントリオール大学、ケベック大学、ドイツのボン大学、ケルン大学、テュービンゲン大学、ベルギーのアントワープ大学、イングランドのノッティンガム大学、ラトビアのリガストラディンス大学、その他オランダ、アメリカ、インド、サウジアラビア等の計10カ国、25施設の共同研究の成果である。我々は、日本人1家系を含む10家系のPIGB欠損症を報告した。PIGBはGPI アンカーの3番目のマンノースを付加する酵素で、その活性低下によりGPIアンカー生合成が減少し細胞膜上の種々のGPI-APの発現が低下する。末梢血のフローサイトメトリー検査での顆粒球上のGPI-AP, CD16の低下の検出が診断に有効で、検査可能であったすべての症例で低下していた。重症のIGDと症状が完全に重複する疾患としてDOORS (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures) 症候群があり、難聴・爪欠損・短い指骨・知的障害/発達遅滞・けいれんを来す。DOORS症候群の責任遺伝子はTBC1D24であるとされているが、今回以前にDOORS症候群と診断された1家系が、PIGB欠損症であることがわかった。TBC1D24の変異を原因とするDOORS症候群はしばしば尿中ケトグルタル酸が異常高値になることが知られているが、このPIGB欠損症の1家系においても同様に異常高値を示しており共通の代謝異常が示唆された。TBC1D24はGPI-APのエンドサイトーシスやリサイクリングに係わっている可能性があり、IGDとDOORS症候群は共通の病態を持っているのかもしれない。
本研究成果は、2019年6月27日にAmerican Journal of Human Genetics誌に掲載されました。
タイトル:Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in the severe cases.
著者:Yoshiko Murakami, Thi Tuyet Mai Nguyen, Nissan Baratang, Praveen K Raju, Alexej Knaus, Sian Ellard, Gabriela Jones, Baiba Lace, Justine Rousseau, Norbert F Ajeawung, Atsushi Kamei, Gaku Minase, Manami Akasaka, Nami Araya, Eriko Koshimizu, Jenneke van den Ende, Florian Erger, Janine Altmüller, Zita Krumina, Jurgis Strautmanis, Inna Inashkina, Janis Stavusis, Areeg El-Gharbawy, Jessica Sebastian, Ratna Dua Puri, Samarth Kulshrestha, Ishwar C Verma, Esther M. Maier, Tobias B Haack, Anil Israni, Julia Baptista, Adam Gunning, Jill A Rosenfeld, Pengfei Liu, Marieke Joosten, María Eugenia Rocha, Mais O. Hashem, Hesham M Aldhalaan, Fowzan S Alkuraya, Satoko Miyatake, Naomichi Matsumoto, Peter Krawitz, Elsa Rossignol, Taroh Kinoshita, Philippe M Campeau