Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation. (Kinoshita Lab, in JCI)
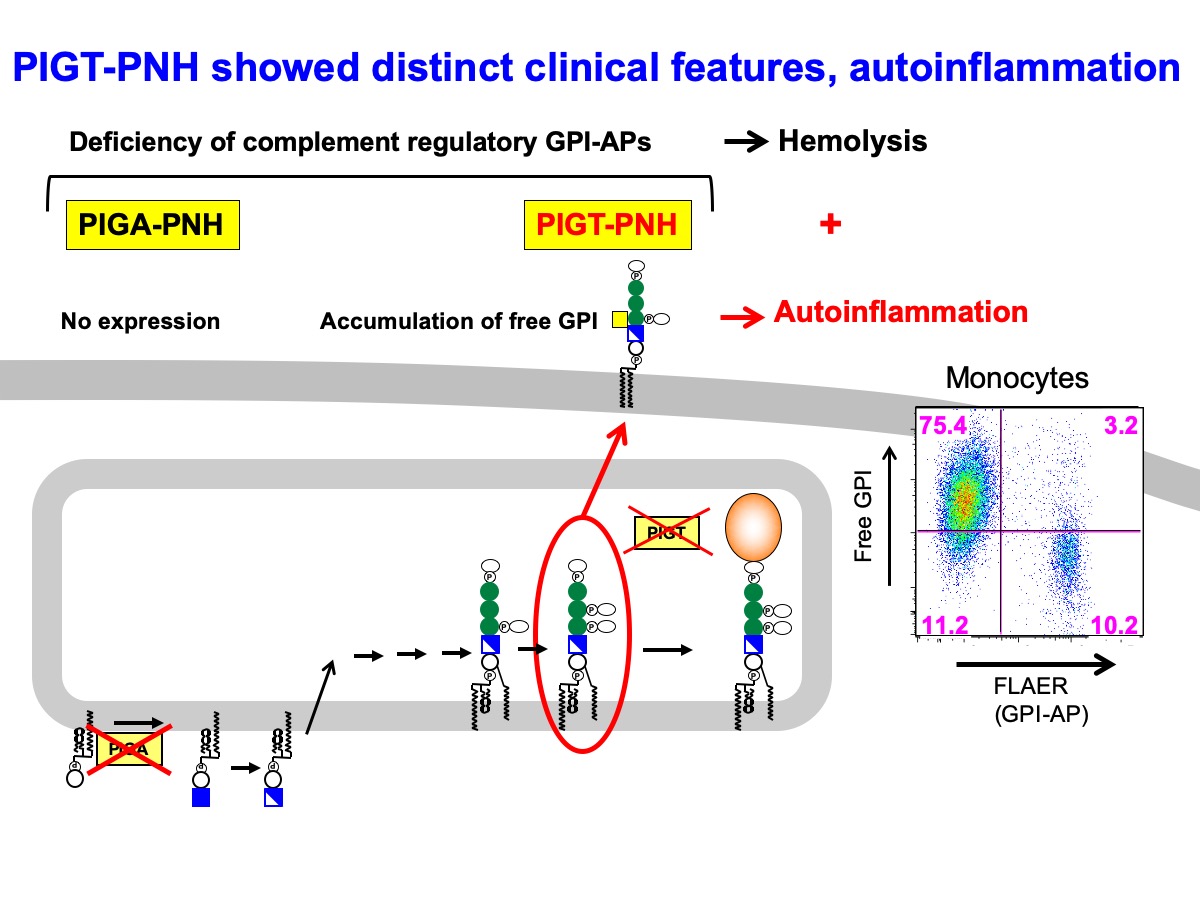
Here, we report the mechanistic basis of atypical paroxysmal nocturnal hemoglobinuria (PNH) caused by germline mutation plus somatic loss of PIGT gene on chromosome 20q, termed PIGT-PNH. PIGT-PNH is characterized by relapsing autoinflammatory symptoms as well as hemolysis. Anti-C5 eculizumab prevented hemolysis and autoinflammation. Because PIGT is required for transfer of synthesized glycosylphosphatidylinositol (GPI) to proteins, PIGT-defective PNH cells lost GPI-anchored complement regulators similar to PIGA-defective PNH cells but also expressed non-protein-linked free GPIs on the cell surface, which does not occur in PIGA-defective cells. Blood leukocytes from a PIGT-PNH patient secreted much higher levels of IL-1b than those from PNH patients after stimulation with inflammasome activators. Studies with PIGT-knockout and PIGA-knockout THP-1 cells demonstrated that complement activation is enhanced on PIGT-knockout cells and higher levels of IL-1b was secreted dependent upon C5b-9 membrane attack complexes. These results suggest a causal role of high free GPIs in increased generation of C5b-9 complexes and IL-1b secretion. Furthermore, a region containing maternally imprinted genes implicated in clonal expansion in 20q- myeloproliferative syndromes was somatically lost together with normal copy of PIGT. Therefore, the mechanism of clonal expansion in PIGT-PNH seems different from that in PNH.
This article was published in The Journal of Clinical Investigation in August 2019.
Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation.
Höchsmann B, Murakami Y, Osato M, Knaus A, Kawamoto M, Inoue N, Hirata T, Murata S, Anliker M, Eggermann T, Jäger M, Floettmann R, Höellein A, Murase S, Ueda Y, Nishimura JI, Kanakura Y, Kohara N, Schrezenmeier H, Krawitz PM, Kinoshita T.
Links
- Home
- Achievement
- Research Activities
- Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation. (Kinoshita Lab, in JCI)






