/Division of Host Defense Laboratory of Immunoglycobiology
Glycosylphosphatidylinositol (GPI) is a glycolipid attached to proteins and anchors them onto the plasma membrane. GPI-anchored protein has various and important physiological functions in our body. Why proteins have this peculiar structure like GPI? Our research goal is to elucidate biogenesis, transport and remodeling of GPI-anchored proteins and understand its physiological significance in our body.
How are GPI-anchored proteins (GPI-APs) regulated?
GPI anchors are synthesized in the endoplasmic reticulum and attached to the C terminus of proteins during posttranslational modification. GPI-anchored proteins are transported from the endoplasmic reticulum to the Golgi and further to the cell surface in a way that is regulated according to the features of GPI. Recently, we identified the enzyme that can cut GPI-anchors, and showed GPI-APs can be secreted and work in the tissues distant from its origin. This result indicates that GPI anchors enable our body system to regulate where and when the protein works in a various way. We are currently studying the molecular mechanism to control the functions of GPI-APs. In addition, GPI anchor has specific carbohydrate side-chains and intriguingly, the chain varies among cells and proteins. We are interested in the physiological significance of this carbohydrate chain and asking how this chain is synthesized in our cells.
Molecular mechanisms of GPI deficiencies.
We found that paroxysmal nocturnal hemoglobinuria (PNH) is caused by somatic mutation of the X-linked PIGA gene, a gene for GPl-anchor biosynthesis. Recently, we reported four cases of the atypical PNH caused by the somatic microdeletion of one allele of the PIGT gene in combination with a germline mutation in the other allele. These patients showed autoinflammatory symptoms in addition to the common symptoms of PIGA-PNH. We are now accumulating similar cases.
We also identified a disease called inherited GPI deficiency (IGD) caused by the mutation of the GPl-anchor synthesizing enzyme, PIGM. The recent whole exome sequencing analysis using the next generation sequencer revealed 24 GPl-related gene mutations responsible for IGD. To elucidate the molecular mechanisms of the pathogenesis of this disease, we developed the system to analyze GPI biosynthesis and modification. This system contributes to the IGD research in all over the world. Our aim is to elucidate how GPl-anchors are involved in IGD and find the way to overcome this disease.
-
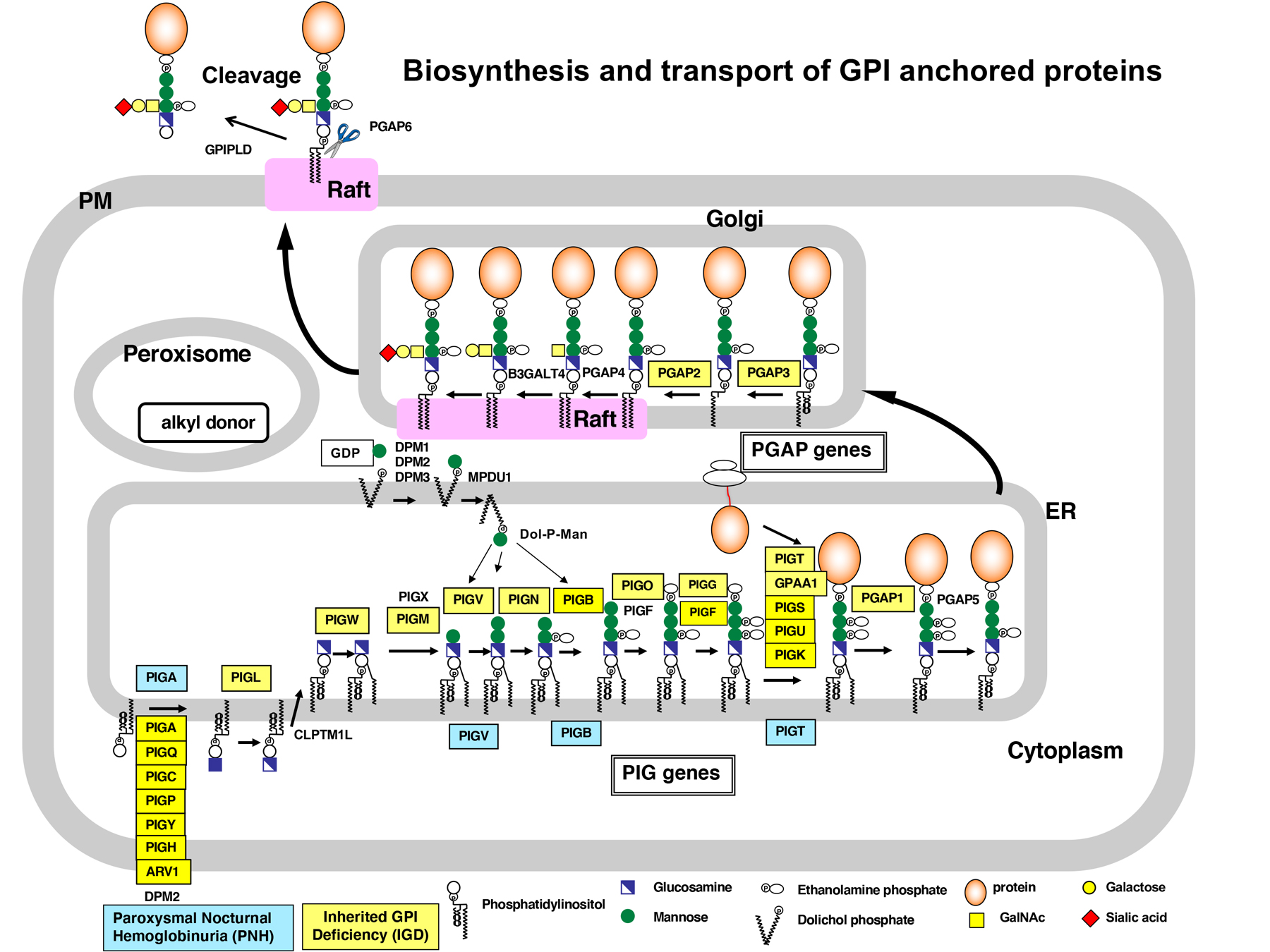
GPI-anchor biosynthesis and the transport/remodeling of GPI-APs.
Staff
- Prof. : Sho Yamasaki (concur.)
- SA Prof. : Yoshiko Murakami
- Postdoc: Saori Umeshita
- Postdoc: Kae Imanishi
Website
Publications
- (1) AAV-based gene therapy ameliorated CNS-specific GPI defect in mouse models. Murakami Y et al.Mol Ther Methods Clin Dev. (2023) 32(1):101176.
(2) Accumulated precursors of specific GPI-anchored proteins upregulate GPI biosynthesis with ARV1. Liu YS et al. J Cell Biol. (2023) 222(5):e202208159.
(3) Genome-wide CRISPR screen reveals CLPTM1L as a lipid scramblase required for efficient glycosylphosphatidylinositol biosynthesis. Wang Y. et al., Proc. Natl. Acad. Sci. USA, (2022) 119(14): e2115083119.
(4) Ethanolamine-phosphate on the second mannose is a preferential bridge for some GPI-anchored proteins. Ishida M., et al., EMBO Rep. (2022) e54352.
(5) Establishment of a mouse model of inherited PIGO deficiency and therapeutic potential of AAV-based gene therapy with genome editing. Kuwayama R., et al., Nat. Commun. (2022) 13:3107.
(6) Sequential hydrolysis of FAD by ecto-5’ nucleotidase CD73 and alkaline phosphatase is required for uptake of vitamin B2 into cells. Shichinohe N., J. Biol. Chem. (2022) 298:102640.
(7) Paroxysmal nocturnal hemoglobinuria caused by CN-LOH of constitutional PIGB mutation and 70-kbp microdeletion on 15q Langemeijer S. et al. Blood Adv. (2020) 4(22):5755-5761.
(8) Cross-talks of glycosylphosphatidylinositol biosynthesis with glycosphingolipid biosynthesis and ER-associated degradation. Wang Y et al. Nat. Commun. (2020) Feb 13;11(1):860.
(9) Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with auto inflammation Hochsmann B. et al. J Clin Invest. (2019) 129 (12):5123-5136.
(10) Mutations in PIGB cause an inherited GPI biosynthesis defect with an axonal neuropathy and metabolic abnormality in the severe cases Murakami Y. et al. (2019) Am. J. Hum. Genet., 105:384-394.
(11) Identification of a Golgi GPI-N-acetylgalactosamine transferase with tandem transmembrane regions in the catalytic domain. Hirata, T ., et al. Nat. Commun. (2018) 9: 405






