Standley Lab/Bioinformatics Center Department of Genome Informatics
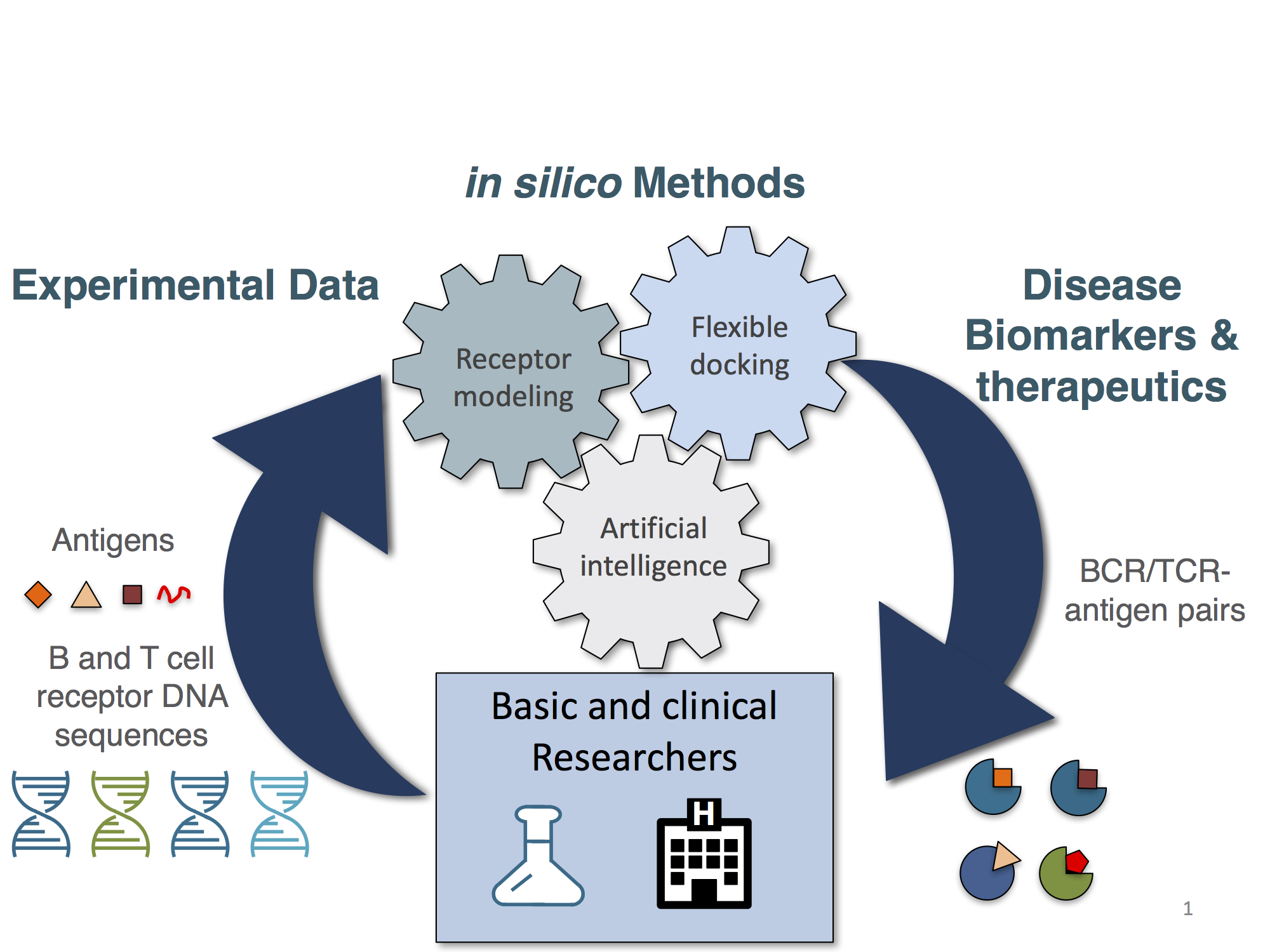
We use single cell sequencing along with computational methods to study problems that are difficult or impossible to observe by experimental methods alone. Some of the problems we work on include: analysis of B and T cell receptor repertoires, protein-nucleotide interactions and multiple sequence alignment of protein and nucleotide sequences. These themes are described in more detail below.
Multiple sequence alignment
Multiple sequence alignment (MSA) is an important step in many computational biology pipelines and MAFFT is one of the most popular programs for building MSAs[1]. Since the first release of MAFFT in 2002, we have been continuously improving its accuracy, speed and utility in practical situations, and have provided different options for newly emerging types of data and analyses. Recent features include: inclusion of secondary structural information of non-coding RNAs and proteins, interactive selection of sequences for phylogenetic tree inference, and integration of protein sequences with comprehensive structural alignments[2]. The latter feature plays a central role in structural modeling methods in our lab.
Analysis of B and T cell receptor specificity and repertoires
Prediction of B and T cell receptor antigen specificities from sequence is currently an important and open problem. Our lab is approaching this challenge using a combination of B and T cell receptor sequencing, structural modeling and artificial intelligence. We have developed a tool for generating BCR and TCR 3D models in a high-throughput and accurate manner[3]. We have further extended this technology to cluster such models according to their antigen and epitope specificity[4]. We have also developed a tool to build TCR-epitope-MHC structural models from sequence[5] and are working on new BCR epitope prediction methods that make use of structural information. The immediate goals of this research are to identify antigens and epitopes that are associated with specific diseases along with the B or T cell receptors that recognize these antigens and epitopes.
Sequencing B and T cell receptors
A healthy immune system maintains a vast repertoire of B and T cells that can recognize a wide range of molecules. B and T cell receptors are produced by the combination of two highly variable polypeptide chains which are encoded by different mRNA transcripts. Due to the high variability of each polypeptide and the combination of two different molecules, each individual’s repertoire of receptors is in general unique. Several immunological and molecular methodologies have been employed to study the immune repertoire with a rather low-resolution. Nevertheless, the advent of next generation sequencing (NGS) has allowed to analyze millions of immune receptor sequences in one sample (bulk sequencing). This has been of great value to the study of immune repertoires, but cannot reveal the pairing of receptor sequences. In the past few years, single cell sequencing technologies have emerged and have made it possible to study paired polypeptide chains from thousands of individual B and T cells. We are currently making use of both bulk and single cell sequencing techniques to study immune cell repertoires in health and disease.
Protein-nucleotide interactions
Protein-nucleotide interactions play a central role in the flow of biological information in all living systems. In the immune system, the importance of DNA-binding proteins in the regulation of transcription has been studied extensively. More recently, the importance of RNA-binding proteins (RBPs) in maintenance of homeostasis as well as in shaping the strength and duration of immune responses post-transcriptionally has been noted[6]. In order to gain further insight into the mechanisms of RBP-mediated immune regulation, we are developing tools for nucleotide binding site prediction and flexible protein-nucleotide docking which have been validated in a number of experimental studies.
Staff
- Prof.: Daron M. Standley
- Assoc. Prof.: Songling Li
- SA Assoc. Prof.: Soyoung Park (concur.)
- Asst. Prof. :. : Arthur R. Millius
- SA Asst. Prof. :. : Hedra Saputra Ismato
Website
Publications
(1) MAFFT multiple sequence alignment software version 7:improvements in performance and usability. Katoh, K.,et al., Mol Biol Evol (2013) 30(4) 772-80
(2) MAFFT-DASH: integrated protein sequence and structural alignment. Rozewick J., et al. Nucleic Acids Research (2019) 47(1)5-10
(3) Repertoire Builder: High-throughput structural modeling of B and T cell receptors. Schritt D., et al. Mol.Syst Des.Eng. (2019) 4,761-768
(4) Functional clustering of B cell receptors using sequence and structural features. Xu Z., et al. Mol. Syst.Des. Eng. (2019) 4, 769-778
(5) Structural Modeling of Lymphocyte Receptors and Their Antigens. Li S., et al. Methods Mol Biol. (2019) 2048:207-229
(6)Regnase-1 and Roquin Regulate a Common Element in lnflammatory mRNAs by Spatiotemporally Distinct Mechanisms.Mino, T.,et al. Cell (2015)161, 1058-1073
- Home
- Laboratories
- Standley Lab






