Hara Lab/Division of Cellular and Molecular Biology Department of Molecular Biology
It has become apparent that aging has a major impact on the incidence of cancers. However, the underlying mechanisms are unclear. We think that cellular senescence plays a key role. In our laboratory, we are aiming to understand the roles and mechanisms of cellular senescence in vivo. We believe that understanding the molecular mechanisms underlying cellular senescence in vivo will provide valuable insight into the development of aging-associated diseases such as cancer, and open up new possibilities for their control.
Exploring the physiological roles and mechanisms underlying cellular senescence in vivo
Cellular senescence is the state of irreversible cell cycle arrest that can be induced by a variety of potentially oncogenic stimuli and has, therefore, long been considered to suppress tumorigenesis. We reported that p16INK4a and p21Waf1/Cip1, both cyclin-dependent kinase inhibitors, play crucial roles in both the onset and establishment of cellular senescence in cell culture and in mouse models. Recently, we generated transgenic mice expressing firefly luciferase under the control of the p16INK4a or p21Waf1/Cip1 gene promoters.
Using these senescence response reporter mice in combination with knockout mice, we are investigating the timing and, hence, the likely roles and mechanisms, of cellular senescence in vivo.
Understanding the molecular mechanisms underlying inflammatory diseases induced by senescence-associated secretory phenotypes (SASPs)
In addition to stable cell cycle arrest, senescent cells also develop senescence-associated secretory phenotypes (SASPs), which contribute both positively and negatively to the onset of inflammatory diseases such as cancer (depending on the biological context). Despite considerable progress in understanding the biological roles of SASPs, far less is known about how they are induced.
Thus, a greater understanding of the underlying molecular mechanisms will lead to novel therapeutic strategies for various aging-associated diseases, including cancer.
Similar to aging, obesity is associated with cancer. However, the underlying mechanisms are not well understood. Recently, we traced the association between obesity and increased cancer risk to gut microbiota communities that produce DNA-damaging bile acid. We found that DNA-damaging bile acid promotes development of obesity-associated liver cancer by inducing SASPs in hepatic stellate cells. We are now focusing on the potential clinical implications of these findings.
-
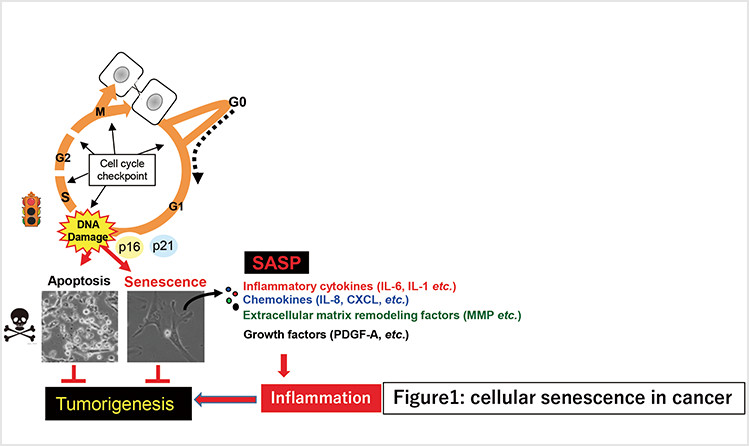
Fig. 1. Cellular senescence initially inhibits proliferation of damaged cells, thereby acting as a fail-safe mechanism. However, in the long term, senescent cells may eventually promote tumorigenesis via SASPs.
-
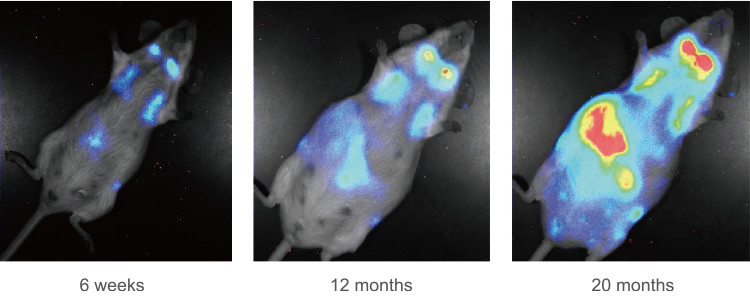
Fig. 2. Real-time bioluminescence imaging of p16INK4a gene expression during aging (Journal of Cell Biology 186: 393-407. 2009).
Staff
- Prof.: Eiji Hara
- Assoc. Prof.: Masahiro Wakita
- Asst. Prof.: Ken Uemura
- SA Asst. Prof.: Yumi Katayama
- SA Asst. Prof.: Noriko Shinjyo
Website
Publications
(1) Comparative analysis of senolytic drugs reveals mitochondrial determinants of efficacy and resistance. Wakita M., et al., Nature Aging (2026) 6: 316-328
(2) Bacterial induction of B-cell senescence promotes age-related changes in the gut microbiota. Kawamoto S., et al., Nature Cell Biology (2023) 25: 865-876
(3) SARS-CoV-2 infection triggers paracrine senescence and leads to a sustained senescence-associated inflammatory response. Tsuji S., et al. Nature Aging (2022) 2: 115-124
(4) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Yoshimoto S., et al., Nature (2013) 499:97-101
(5) DNA damage signaling triggers degradation of histone methyltransferases through APC/CCdh1 in senescent cells. Takahashi A., et al., Molecular Cell (2012) 45:123-31
- Home
- Laboratories
- Hara Lab






