バイオインフォマティクスセンター 生物情報解析分野 ゲノム進化医科学グループ/中谷研究室
ゲノムは生命の進化を記録した歴史書とも言われます。この「進化の歴史書」から、私たちはどのような情報を読み解くことができるのでしょうか。そして、ゲノムに刻まれた進化の記録は、ヒトの遺伝性疾患やがんの発症要因となるゲノム異常の理解にどのように貢献するのでしょうか。こうした問いに答えるため、ゲノム進化医科学グループでは、進化遺伝学・腫瘍学・バイオインフォマティクスなどの学際的アプローチを駆使し、ゲノム情報解析・マルチオミクス解析・アルゴリズム開発に取り組んでいます。ゲノム進化の研究を通じて、遺伝子やゲノムの働きをより深く理解し、がんゲノム進化との関連や遺伝性疾患の発症メカニズムの解明へとつなげることを目指しています。
研究テーマは多岐にわたりますが、主要な研究としては以下のような進化ゲノム学研究やがんゲノム解析が挙げられます。
ゲノム構造進化の確率モデル構築によるメダカのゲノム進化史の解明
哺乳類・鳥類・魚類といった遠縁脊椎動物ゲノムが次々と解読されることで、5億年にわたる脊椎動物の進化がゲノムレベルで議論できるようになりました。しかし、魚類をはじめとする遠縁の脊椎動物間では遺伝子の並びが大きく変化しており、進化の過程で生じたゲノム構造の変化を追跡することは困難でした。この課題に取り組むため、私たちは染色体レベルの遺伝子構成に着目し、ゲノム構造進化の推定手法を研究してきました。論文1では、メダカゲノムを解読し、ヒトやミドリフグのゲノムと比較することで約3.5億年間にわたる真骨魚類のゲノム構造進化を推定しました。その結果、ゲノム構造の進化は単調なものではなく、比較的短期間に大規模な変化が生じていたことが明らかになりました。さらに、論文4ではゲノム構造進化を確率モデルとして定式化し、祖先ゲノム構造を推定するベイズ推定アルゴリズムを開発しました(図1)。「ゲノムは生命進化の歴史書である」とよく表現されますが、この「歴史書」に対してテキスト解析分野で使われる「トピック構造」の推定アルゴリズムを応用することで「祖先ゲノム構造」の推定が可能になり、ゲノム進化モデリングの研究として高く評価されました。その後、この手法をゾウギンザメやヤツメウナギのゲノム解析にも応用し、初期脊椎動物のゲノム進化の研究にも取り組んでいます。
5億年前の初期脊椎動物進化におけるゲノム倍数化イベントの研究
無脊椎動物から脊椎動物へと進化する過程で、ゲノム全体が2回重複したという仮説が古くから注目されていました。しかし、5億年前に起きた全ゲノム重複を検証することは困難であり、さらに脊椎動物の祖先ゲノム構造に関する手掛かりはほとんど存在しない状況でした。そこで私たちは、全ゲノム重複の証明を目指し、脊椎動物の祖先ゲノムを推定する解析手法を研究してきました。論文2では、ゲノム中の重複遺伝子の「分布」に着目することで全ゲノム重複後の染色体構造を再構成する手法を考案しました。その結果、初期脊椎動物の系統において全ゲノム重複が2回起きてゲノム全体が2×2=4倍になっていることが確認されました。さらに、論文5ではこの解析手法を応用し、ヒトから最も遠縁の脊椎動物であたる円口類の一種であるカワヤツメのゲノムを解析しました。その結果、再構成された円口類祖先ゲノムは、従来想定されていた2×2=4倍や2×2×2=8倍ではなく、2×3=6倍となっていることが判明しました。これにより、それまで知られていなかった円口類祖先の全ゲノム3倍化現象が発見されました(図2)。これら複数回のゲノム倍数化を通じて複雑な個体発生システムや獲得免疫システムが進化したと考えられています。私たちはこうした複雑な生命システムを生み出したゲノム進化メカニズムを解明することを目指し、研究を進めています。
がんゲノムのオミクスデータ解析と空間的遺伝子発現解析によるクローン進化の研究
5億年前に起きた全ゲノム重複の研究は、ヒトゲノムへの理解を深める上でどう役立つのでしょうか? これまでの研究により、5億年前の全ゲノム重複で生まれた重複遺伝子はダウン症候群をはじめとするヒトの遺伝性疾患と関連していることがわかってきています。また、「がんはゲノムの病気」と言われるように、体細胞のゲノムに変異が蓄積することでがんは発症します。特に、がん細胞のゲノムには全ゲノム重複や染色体コピー数異常などの大規模なゲノム構造異常が多く見られることが知られています。そこで私たちは、生物のゲノム進化とがん細胞のゲノム進化の関連性を解明することを目標に、がんゲノムの構造解析を進めています。論文6では、膵神経内分泌腫瘍の腫瘍形成過程において、特定の染色体が欠失しやすいことや、全ゲノム重複が多くの症例に生じていることを明らかにしました。さらに、このような染色体コピー数異常による発症メカニズムを理解するため、空間的遺伝子発現データを収集し、腫瘍形成過程におけるクローン進化の研究を進めています(図3)。
上述のように、私たちは計算機科学・確率統計学の専門性を生かし、多様な生命医科学データを解析しています。バイオインフォマティクスの研究室として情報解析や数理解析を重視していますが、微生物病研究所や医歯薬生命系研究科との共同研究により新規データ解析にも取り組んでいます。最先端データの解析を通じて、ゲノム進化学とゲノム医科学との融合領域でのバイオインフォマティクス研究を推進したいと考えています。
-
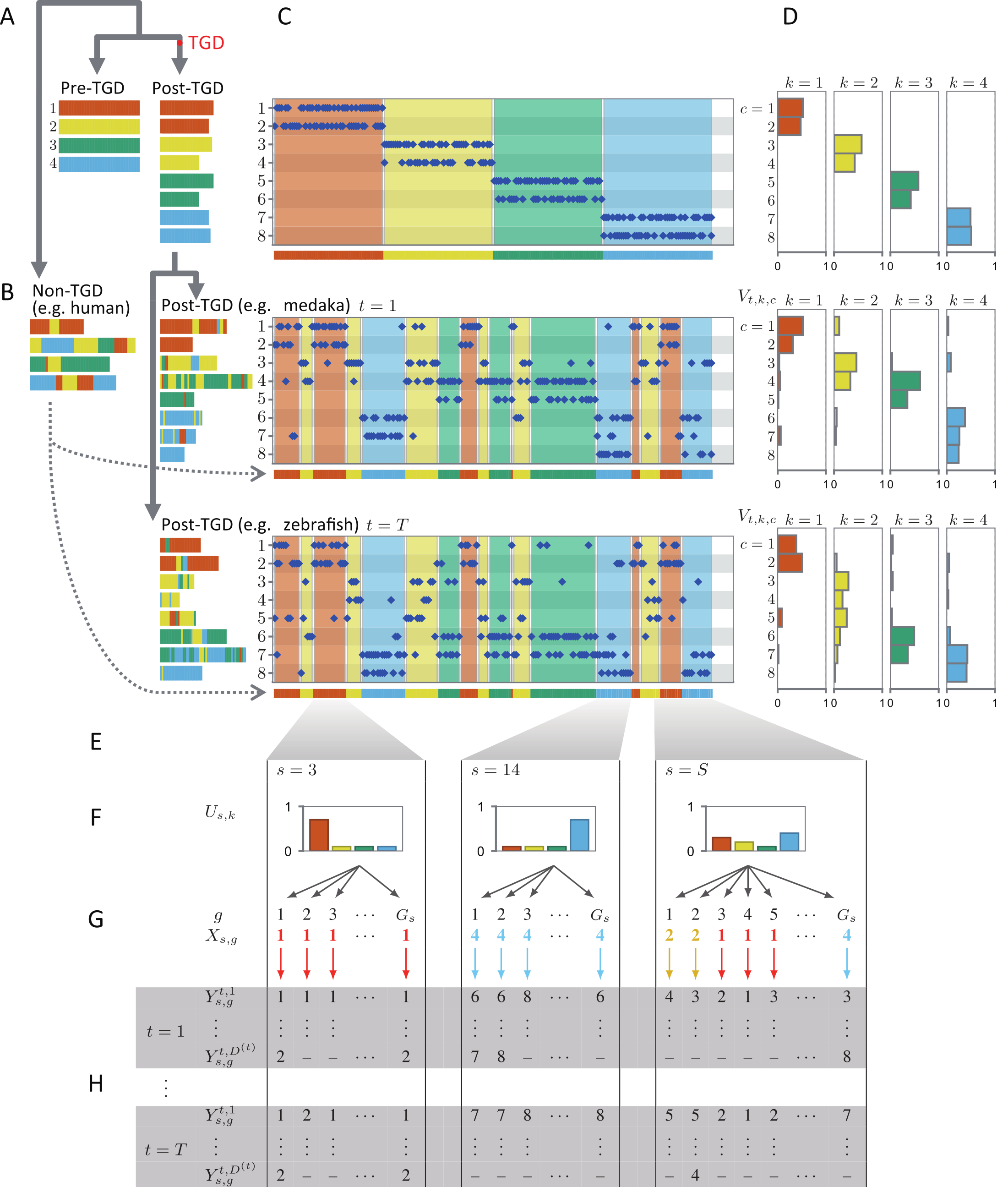
図1. ゲノム構造進化の確率モデル. 染色体レベルの遺伝子構成進化の確率モデルを考え、変分ベイズ推定アルゴリズムを適用することで祖先ゲノム構造を推定する手法を開発した。この手法により、全ゲノム重複が起きた種に対して祖先ゲノムの高精度な推定が可能になった。(確率モデルと図の詳細については論文4を参照)
-
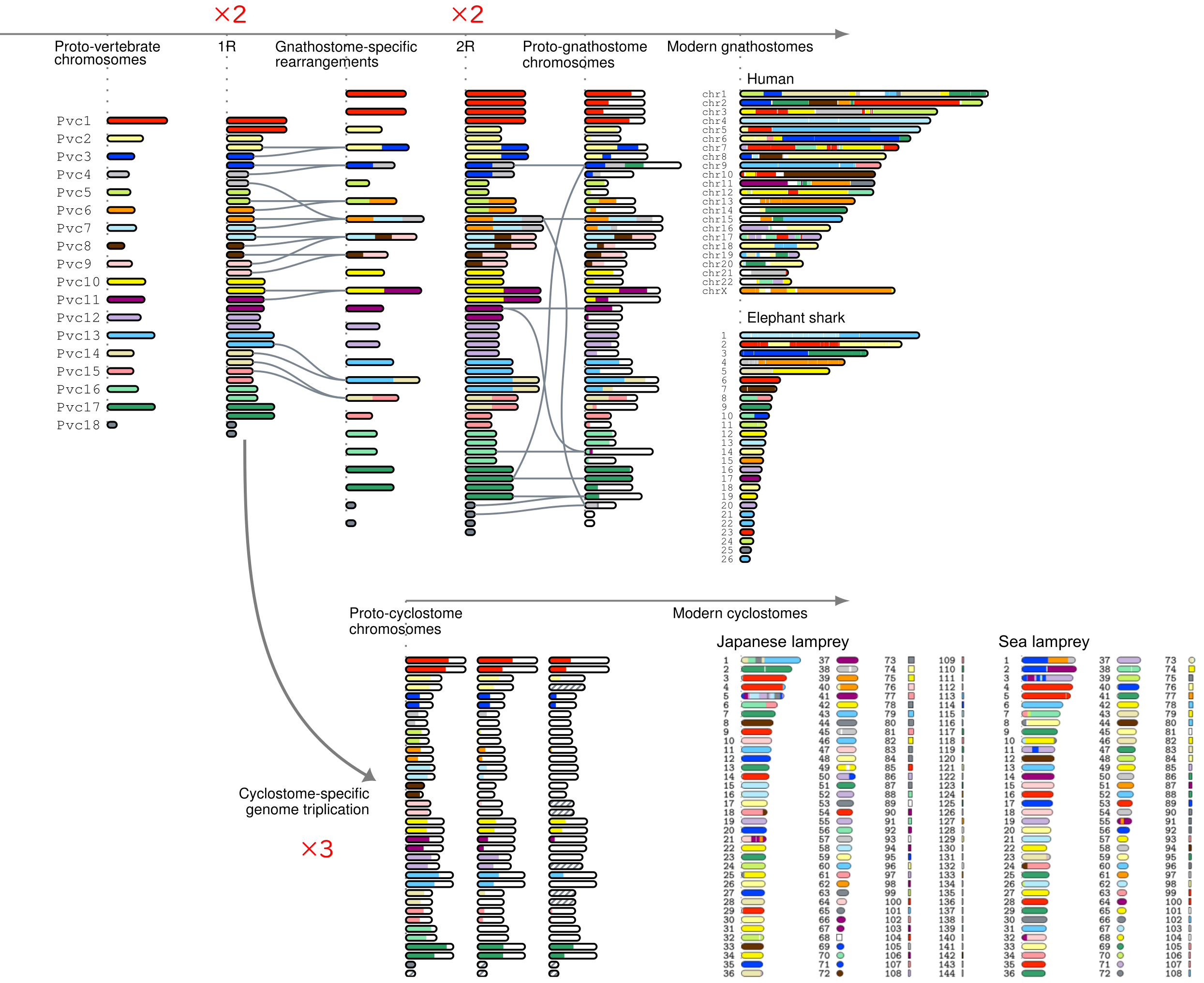
図2. 初期脊椎動物のゲノム進化. カワヤツメ(Japanese lamprey)とゾウギンザメ(elephant shark)のゲノム解読により初期脊椎動物のゲノム進化史が明らかになった。脊椎動物祖先と顎口類祖先で全ゲノム重複が1回ずつ起き、その結果顎口類祖先ゲノムは4倍(×2×2)になった(上段)。初期円口類系統では、脊椎動物祖先の全ゲノム重複に続いてゲノム全体が3倍化した結果、合計6倍(×2×3)になったことがわかった(下段)。
-
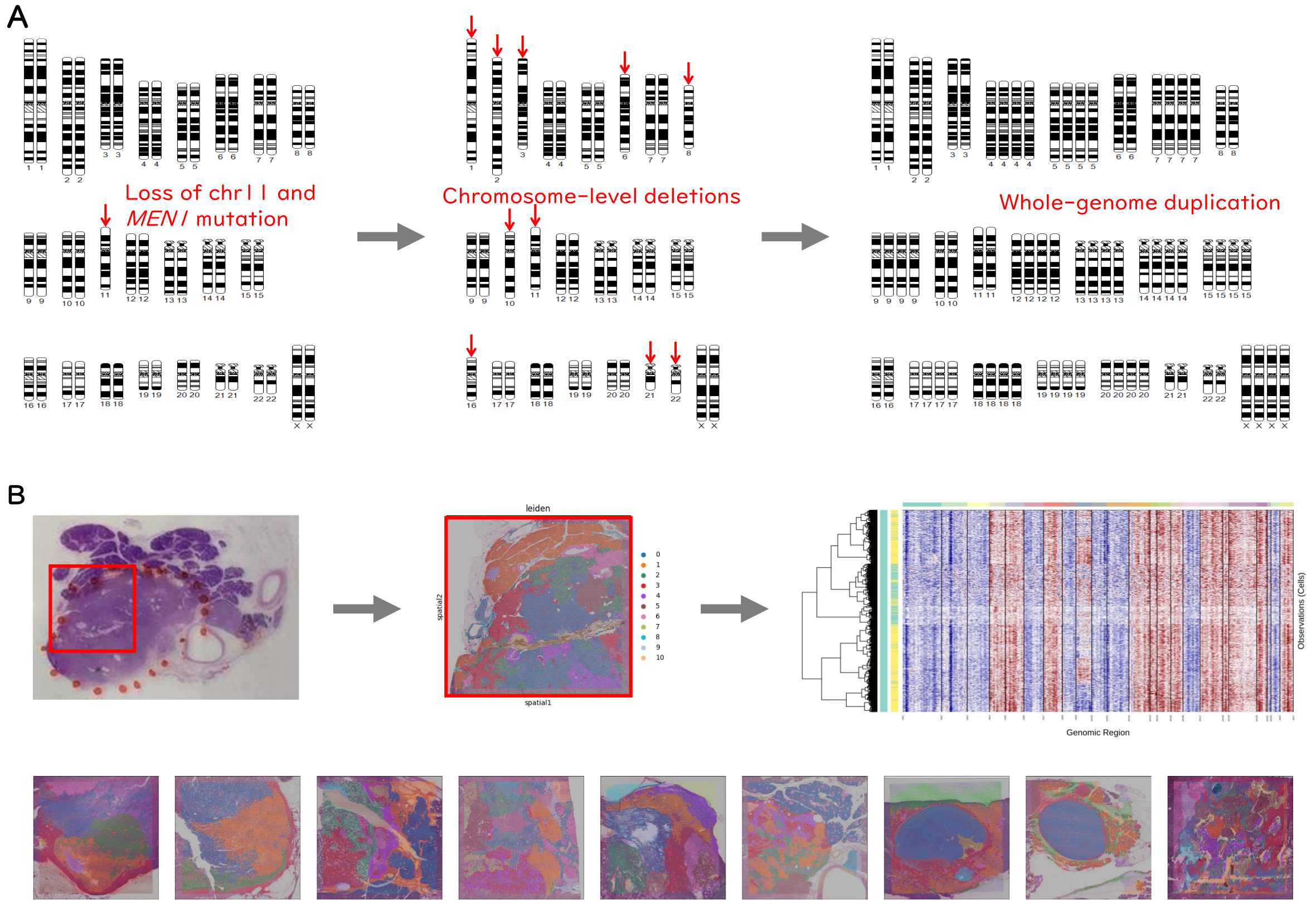
図3. 空間的遺伝子発現データを用いたクローン進化解析. (A) 全ゲノムシークエンスデータのコピー数解析により、膵神経内分泌腫瘍に特有の染色体欠失パターンが存在することが明らかになった。これらの染色体の欠失と全ゲノム重複が膵神経内分泌腫瘍の発症に寄与すると考えられる。 (B) そこで空間的遺伝子発現データを用いたクローン進化解析により、染色体欠失パターンの形成機構の解明に取り組んでいる。
メンバー
- 教授: 中谷 洋一郎
最近の代表的な論文
(1) The medaka draft genome and insights into vertebrate genome evolution. Kasahara, M., et al. Nature (2007) 447, 714-719.
(2) Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Nakatani, Y., et al. Genome Res (2007) 17, 1254-1265.
(3) Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Sasaki, S., et al. Science (2009) 323, 401-404.
(4) Genomes as documents of evolutionary history: a probabilistic macrosynteny model for the reconstruction of ancestral genomes. Nakatani, Y. & McLysaght, A. Bioinformatics (2017) 33, i369-i378.
(5) Reconstruction of proto-vertebrate, proto-cyclostome and proto-gnathostome genomes provides new insights into early vertebrate evolution. Nakatani, Y., et al. Nat Commun (2021) 12, 1-14.
(6) Comprehensive genomic profiling of neuroendocrine carcinomas of the gastrointestinal system. Yachida, S. et al. Cancer Discov (2022) 12, 692-711.






