Nakatani Lab/Bioinformatics Center Department of Biological Informatics Laboratory of Medical and Evolutionary Genomics
Medical evolutionary genomics is a research field that examines genome evolution and its relationship with human health and disease. By integrating expertise in evolutionary genetics, oncology, and bioinformatics, we develop innovative methods to analyze genomic data and identify genetic causes of diseases. In particular, we apply advanced data science techniques, such as probabilistic modeling, machine learning, and artificial intelligence, to detect evolutionary patterns in large-scale genomic datasets. By examining genome evolution, we aim to uncover the mechanisms underlying human genetic disorders and cancer, with the goal of developing novel diagnostic and therapeutic strategies.
Our research covers topics from evolutionary genomics to cancer genome analysis, as described below.
Elucidating the evolutionary history of the medaka genome by developing a probability model of genome structure evolution
Sequencing the genomes of distantly related vertebrates, such as mammals, birds, and fish, has enabled genomic-level studies of vertebrate evolution. However, tracing genomic changes over 500 million years is challenging owing to extensive gene order rearrangements among distantly related species. To address this, we are developing computational approaches that reconstruct genome structure evolution by focusing on chromosome-level gene organization. In [1], we sequenced the medaka genome and compared it with the genomes of humans and green spotted pufferfish, allowing inference of teleost genome evolution over 300 million years. Our findings revealed that genome structure evolution occurred through rapid, large-scale changes rather than gradual alterations. In [4], we modeled genome structure evolution as a probabilistic process and developed a variational Bayesian inference algorithm to reconstruct ancestral genome configurations (Fig. 1). This research demonstrates that genomes can be treated as evolutionary documents: by applying a topic inference algorithm, commonly used in text analysis, we successfully inferred the ancestral genome structure, similar to inferring topic structures from documents.
Elucidating whole-genome duplication and triplication events that occurred during early vertebrate evolution 500 million years ago
It has been hypothesized that two rounds of whole-genome duplication occurred during the evolutionary transition from invertebrates to vertebrates, facilitating increased morphological complexity. However, validating genomic changes from 500 million years ago is difficult owing to limited evidence of ancestral genome structure in early vertebrate lineages. To investigate this, we have developed methods to reconstruct ancestral vertebrate genomes, aiming to validate ancient whole-genome duplication (WGD) events. In [2], we created a computational method to infer post-WGD genome structure by analyzing duplicated gene distributions in the human genome. Our results confirmed two WGD events in early vertebrate evolution, producing a 2 × 2 = 4-fold whole-genome duplication. In [5], we applied this method to the Japanese lamprey genome, a cyclostome species distantly related to humans. The reconstructed protocyclostome genome revealed a 2 × 3 = 6-fold duplication, rather than the previously hypothesized 2 × 2 = 4-fold or 2 × 2 × 2 = 8-fold duplications. This finding uncovered a previously unknown whole-genome triplication event in the early cyclostomes (Fig. 2). These polyploidization events contributed to the emergence of complex developmental pathways and adaptive immune systems. We are also investigating the genetic mechanisms that enabled these biological innovations during and after polyploidization.
Multiomics and spatial transcriptomics analysis of clonal evolution during tumorigenesis
How can understanding WGDs from 500 million years ago improve our knowledge of the human genome? Research has shown that cancer genomes frequently undergo large-scale structural changes, including WGDs and chromosomal copy-number aberrations. To explore the link between organismal and cancer genome evolution, we have conducted structural analyses of cancer genomes. In [6], we identified distinct patterns of chromosomal copy-number aberrations, along with frequent WGDs, during the development of pancreatic neuroendocrine tumors. To better understand the disease mechanisms driving these alterations, we are using spatial transcriptomics to investigate clonal evolution during tumorigenesis (Fig. 3).
As outlined above, we have analyzed diverse biomedical data using our expertise in informatics and statistics. As a bioinformatics laboratory, we primarily focus on computational and mathematical analyses; however, we also collaborate on data analysis with the Research Institute for Microbial Diseases and the University of Osaka. Through analysis of cutting-edge data, we aim to advance bioinformatics at the intersection of evolutionary genomics and medical genome science.
-
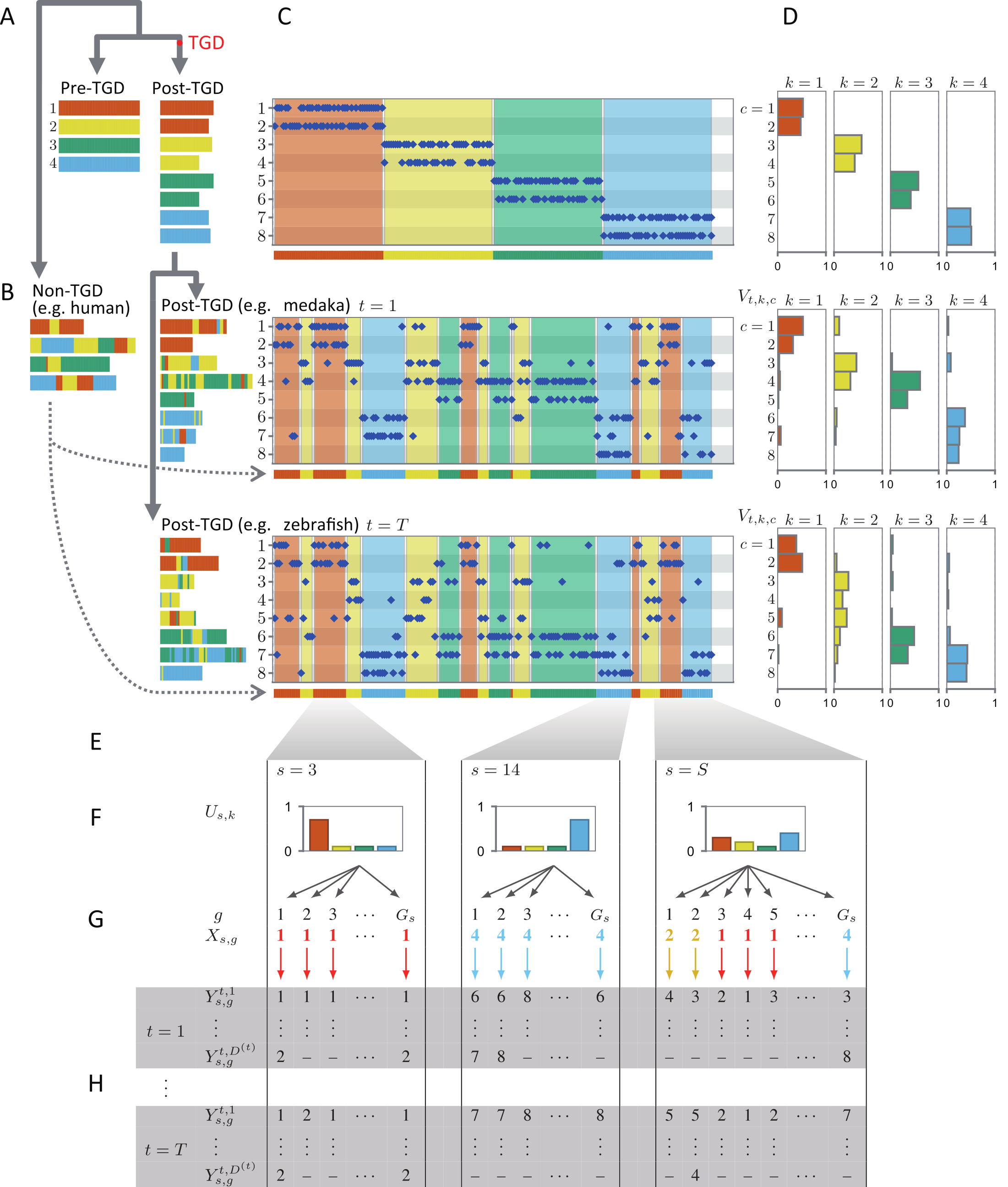
Figure 1. Probabilistic model of genome structure evolution. We developed a probabilistic framework to model the evolution of gene distribution at the chromosome level. Using a variational Bayesian inference algorithm, this approach enabled accurate reconstruction of ancestral genomes in species that experienced whole-genome duplication. Refer to [4] for model details.
-
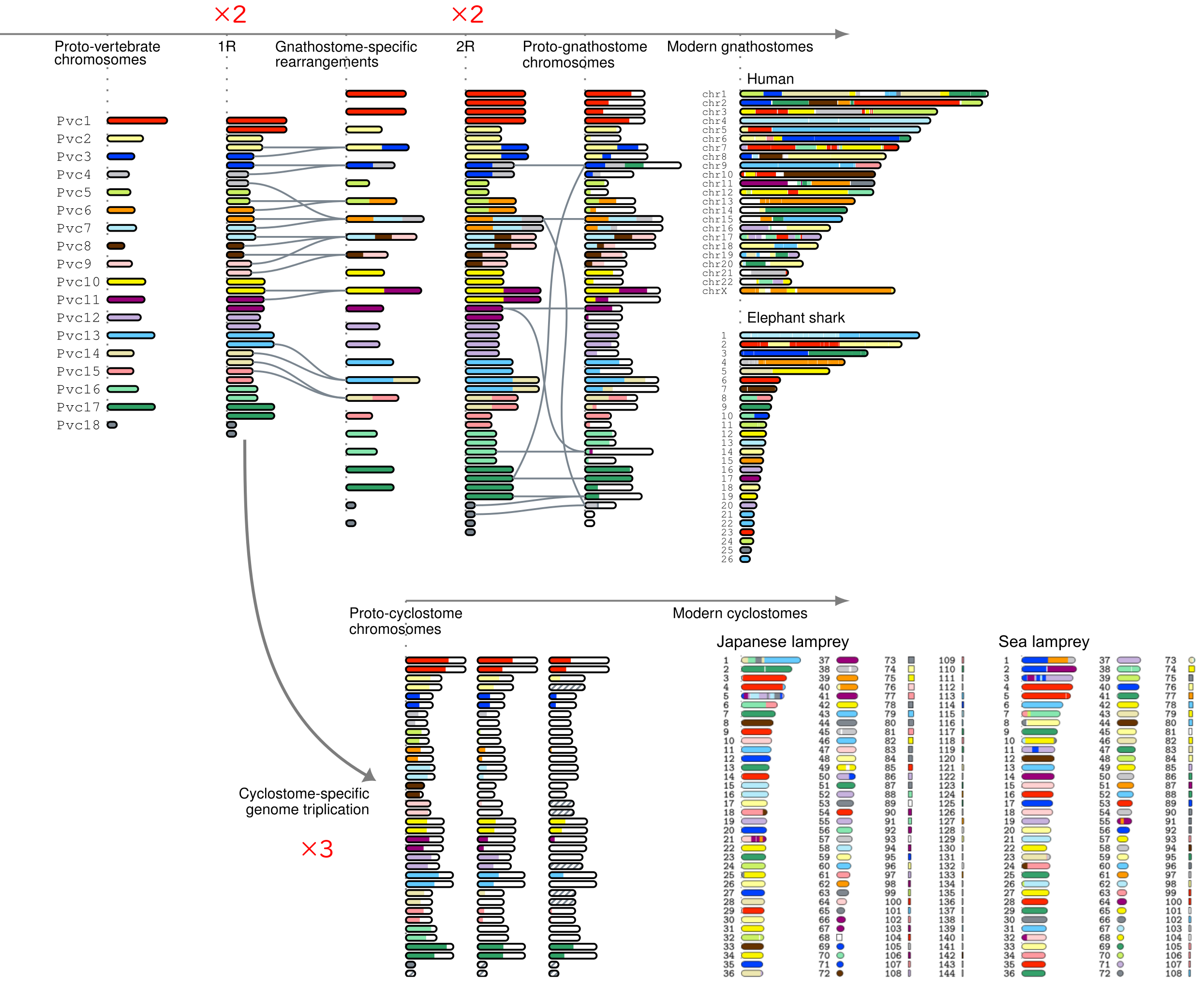
Figure 2. Evolutionary history of early vertebrate genomes. Genome sequencing of the Japanese lamprey and the elephant shark has revealed insights into the evolution of early vertebrate genomes. Whole-genome duplication (WGD) occurred once in the vertebrate ancestor and once in the jawed vertebrate ancestor, leading to a fourfold (2 × 2) genome expansion in the jawed vertebrate lineage (upper panel). In the early cyclostome lineage, an initial WGD in the vertebrate ancestor was followed by a whole-genome triplication, resulting in a sixfold (2 × 3) increase in genomic content (lower panel).
-
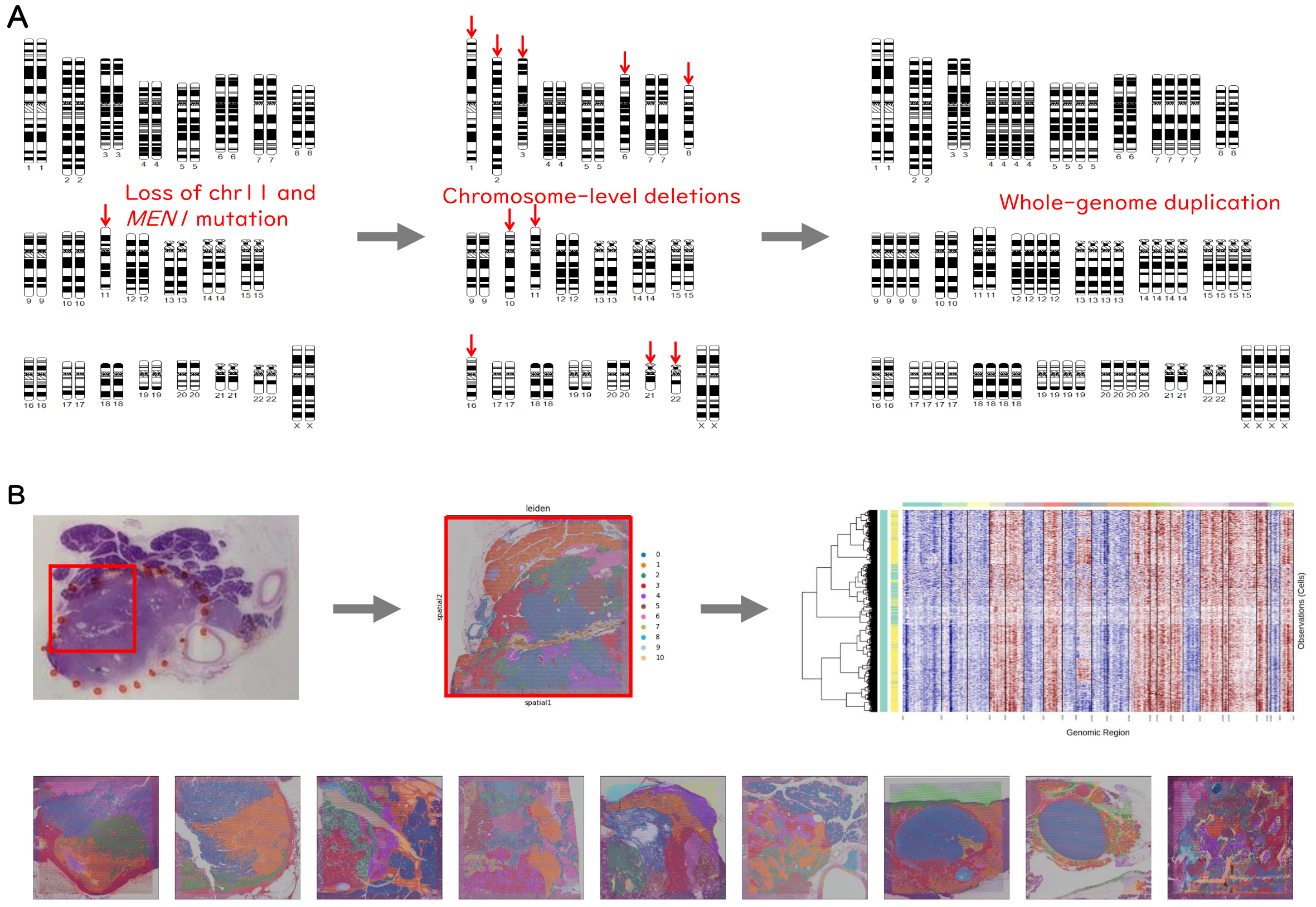
Figure 3. Analysis of clonal evolution using spatial transcriptomics data. (A) Whole-genome sequencing revealed characteristic chromosomal copy-number alterations in pancreatic neuroendocrine tumors. These deletions, along with whole-genome duplications, likely contribute to tumorigenesis. (B) To investigate the mechanisms underlying chromosomal deletions during tumorigenesis, we are analyzing clonal evolution using spatial transcriptomics data.
Staff
- Prof.: Yoichiro Nakatani
Publications
[1]The medaka draft genome and insights into vertebrate genome evolution. Kasahara, M., et al. Nature (2007) 447, 714-719.
[2] Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Nakatani, Y., et al. Genome Res (2007) 17, 1254-1265.
[3] Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Sasaki, S., et al. Science (2009) 323, 401-404.
[4] Genomes as documents of evolutionary history: a probabilistic macrosynteny model for the reconstruction of ancestral genomes. Nakatani, Y. & McLysaght, A. Bioinformatics (2017) 33, i369-i378.
[5] Reconstruction of proto-vertebrate, proto-cyclostome and proto-gnathostome genomes provides new insights into early vertebrate evolution. Nakatani, Y., et al. Nat Commun (2021) 12, 1-14.
[6] Comprehensive genomic profiling of neuroendocrine carcinomas of the gastrointestinal system. Yachida, S. et al. Cancer Discov (2022) 12, 692-711.
- Home
- Laboratories
- Nakatani Lab






