PNHの病態と責任遺伝子
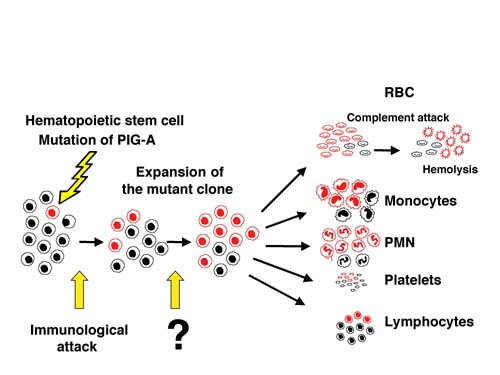
PNHは正常では赤血球膜上に存在する補体制御因子が欠損しているために、自己赤血球が補体により破壊されて、溶血性貧血をきたす血液疾患である。この補体制御因子であるDAF/CD55、 CD59の両因子はglycosyl phosphatidyl inositol anchor (GPI アンカー)と呼ばれる共通の膜結合部分を持ち、異常赤血球はこの部分の欠損によって両因子をはじめとするすべてのGPIアンカー型蛋白の発現を欠く。我々は1993年にGPIアンカー生合成に関わるX染色体上の遺伝子PIG-Aをクローニングし、このPIG-AがPNHにおけるGPIアンカー欠損の責任遺伝子であることを報告した。実際、現在までに解析されたすべてのPNH患者においてPIG-Aの異常が見つかっている。GPIアンカー型蛋白の欠損は赤血球だけではなくすべての血球系においてみられ、また一方では正常の血液細胞もみられる。即ちPNHは後天的にある1つの 造血幹細胞のPIG-A遺伝子に突然変異が起き、GPIアンカー型蛋白が陰性になった後、何らかのメカニズムでクローン性に拡大することにより生ずる疾患といえる。
異常細胞のクローン性拡大のメカニズム
マウスモデルの解析により、PIG-A変異のみではクローンが優位性を獲得することはなく、他の要因が必要であることがわかった。我々はPNHがしばしば再生不良性貧血と併発することから第二の要因として自己免疫的機序を想定し、GPI欠損細胞がその攻撃から免れて選択されうることをマウスモデルを用いて証明した。PNH患者の免疫学的な解析と上記の結果より、現在ではすべての症例に自己免疫的な機序が関わっていると考えられている。
今後の展望
まず自己免疫的機序については、実際に患者において関わっているGPIアンカー型蛋白の同定、認識される抗原と免疫担当細胞の同定は残された課題であり、検体が得難いだけに難しいテーマである。またPIG-Aの突然変異と自己免疫的機序の2ステップですべてのPNHの発症が説明できるかは未だ疑問である。というのは多くの再生不良性患者の末梢血中にはPNHクローンが確認されるにもかかわらず、それらは大多数それ以上増加することなくPNHとして発症しないからである。症例のなかには良性腫瘍化をおこすような第三のステップが関与している可能性があり、我々はこれの追求によりPNH発症機序の全容解明を目指している。