|
|
||
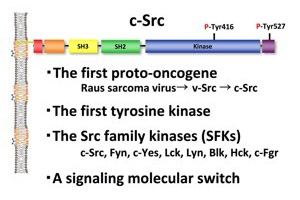
図1.Srcの構造。Srcは最初に同定されたがん遺伝子産物であり、最初のチロシンキナーゼでもある。 Srcはファミリー(SFK)を形成し、正常細胞ではシグナル伝達の分子スイッチとして機能する。 |
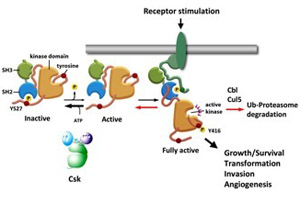
図2.Srcの活性調節機構。Srcは通常、C末端の制御部位(Y527)がCskによってリン酸化された不活性型で存在する。Y527が脱リン酸化されると活性型となるが、その状態ではまだ機能的ではない。細胞が増殖因子などの刺激を受けると、活性化した受容体やアダプターとSrcは結合し、自己リン酸化部位がリン酸化されてフルに活性化して機能する。活性化したSrcはCskによってリン酸化されて再び不活性型にもどるか、あるいはユビキチン化されて分解される。 |
||
 |
 |
||
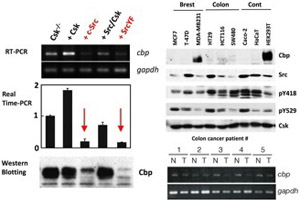
図3.Cbpの遺伝発現がSrcによってがん化した細胞や、Srcの活性が高いヒトがん細胞および組織において著明に低下している。 |
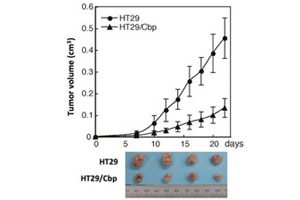
図4.ヒト大腸がん細胞HT29にCbpを発現させるとヌードマウス皮下における造腫瘍活性が抑制される。 |
||
 |
|
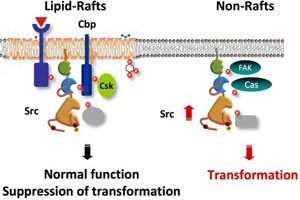
図5.SrcはラフトにCbpを介してトラップされるとその形質転換活性を失う。ラフトの外で異常に活性化することによってSrcは形質転換活性を獲得する。 |
|
 |
 |
||
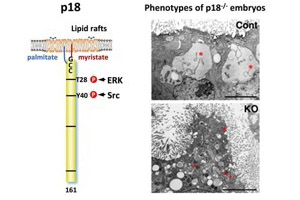
図6.p18の構造とp18欠損マウスの表現型。p18は、N末端の脂肪酸を介して後期エンドソームのラフトに局在化する。SrcおよびERKによるポテンシャルなリン酸化サイトが存在する。p18欠損マウスは胎生致死となる(胎生7日目頃)。近位内胚葉細胞の細胞内膜系(エンドソームやリソソーム)の配置や成熟に大きな異常が観察されている。 |
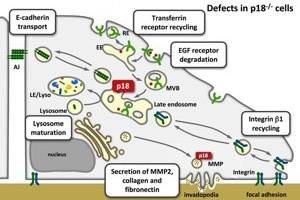
図7.p18欠損細胞ではここに示した様々なエンドソーム系反応において異常が認められる。 |
||
 |
|
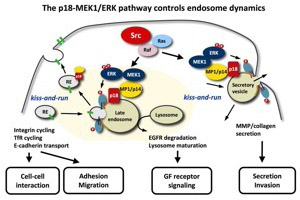
図8.p18の機能に関する作業仮説。p18欠損細胞の形質が、SrcやRasにより形質転換した細胞の形質と強く関連することから、それらの形質転換においてp18を経由するMAPキナーゼ経路が使われる可能性が示唆されている。 |
|
 |
|
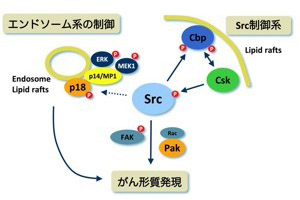
図9.当研究室における研究MAP(作業仮説)。Srcを中心とした細胞形質の制御機構、およびその破綻によるがん化(がん形質発現)機構の解明を目指した研究を進めている。 |
|