Research Projects
Cancers arise, evolve and develop progressively due to the accumulations of mutations and/or modifications in the genomic DNA. Loss-of-function mutations in “tumor suppressor genes” induce cell immortalization, while gain-of-function mutations in “proto-oncogenes” induce cell transformation. Cell immortalization prevents the induction of apoptosis and/or senescence, which is a defense mechanism against cancer development. Cell transformation involves the gain of autonomous cell growth, the loss of cell communication, morphological changes, and the elevated production of matrix proteases and growth factors that participate in invasion, metastasis and angiogenesis. These cellular events thus lead to the malignant conversion of cancer cells. The primary focus of this department is to understand the molecular basis of the cell transformation that is induced by the gain-of-function mutations of proto-oncogenes. As a representative proto-oncogene, we have focused on the c-Src proto-oncogene, which encodes a non-receptor tyrosine kinase. To date, we have analyzed its physiological roles in development and the mechanisms by which its specific regulators, such as Csk and Cbp, regulate it. To obtain a full picture of the cell signaling pathways that lead to c-Src–mediated cell transformation and to search for new therapeutic targets that will block c-Src–mediated cancer progression, the following projects are currently in progress:
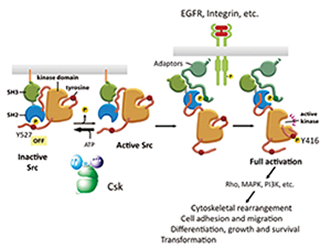 |
Fig. 1. Function and regulation of c-Src. |
I. Function and regulation of c-Src.
In normal cells, c-Src is present as an inactive form that is phosphorylated by its negative regulatory kinase Csk. Extracellular stimuli transiently activate it, after which it in turn activates downstream components such as the MAPK pathway and Rho family GTPases, thereby inducing gene expression and cytoskeletal rearrangements, respectively. This promotes the cell growth and phenotypic changes that are involved in cell transformation (Fig. 1). While the c-SRC gene is rarely mutated in human cancers, its protein is frequently hyperactivated and overexpressed. This aberrant activation of c-Src is suggested to contribute to cancer malignancy. Recently, we showed that the oncogenic potential of c-Src is regulated by the membrane adaptor protein Cbp, which is exclusively localized to the membrane microdomain. We are currently analyzing the relationship between the disruption of this regulatory system and cancer progression.
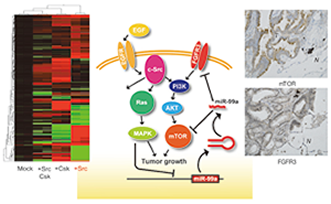 |
Fig. 2. A mechanism of c-Src-induced cell transformation regulated by miRNA. |
II. Mechanism of cell transformation induced by c-Src activation.
To define the signaling pathway that leads to c-Src-mediated cell transformation, we performed a comprehensive study of target molecules by using a newly developed c-Src inducible system. To date, proteomic analyses have found that Arhgef5 is a critical c-Src target. Arhgef5 is a Dbl family of GEF for Rho GTPase. A miRNA array analysis also revealed that c-Src activation alters the gene expression of a limited number of miRNAs. We identified several target genes for these miRNAs and analyzed their functions. Based on the finding that many of the target molecules (e.g., FGFR3, mTOR and ILK) are components of the c-Src signaling pathway, we proposed a new tuning system of c-Src-mediated cell transformation via miRNA. Analysis of the regulatory mechanism for c-Src-mediated miRNA expression is currently in progress.
III. Regulation of cell growth signaling via the late endosome/lysosome.
During the search for c-Src targets, we recently found a new regulator of cell growth signaling, namely, p18, which is a membrane adaptor protein that localizes exclusively to late endosomes/lysosomes. p18 forms a ternary complex with the scaffolding p14/MP1 proteins and plays an essential role in the activation of mTORC1 on late endosomes/lysosomes. p18 KO mice are embryonic lethal and exhibit severe defects in lysosome biogenesis (Fig. 3). To elucidate the function of p18 in lysosome biogenesis, we are currently searching for target molecules for the p18-mTORC1 pathway in late endosomes/lysosomes. We also found that the p18-dependent pathway is crucial for controlling cell growth and oncogene-mediated cell transformation. The elucidation of the underlying mechanism is an important objective of our project as well.
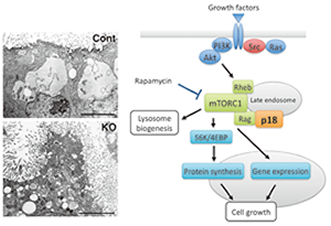 |
Fig. 3. Roles played by the late endosome/lysosome-anchored p18-mTORC1 pathway. |