Research Group
| Professor (SUP) | Taroh Kinoshita |
| Associate Professor | Yusuke Maeda |
| Associate Professor (SUP) | Yoshiko Murakami |
Research Projects
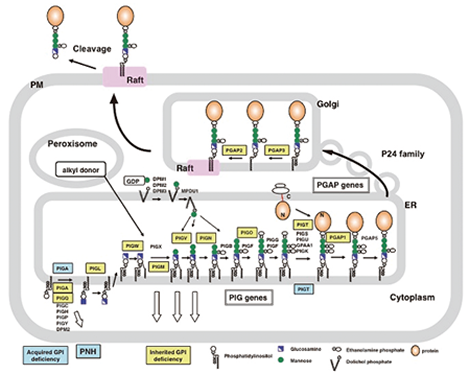 |
Fig. 1 GPI-anchor biosynthesis and the transport/remodeling of GPI-APs. |
Glycosylphosphatidylinositol (GPI) is a glycolipid that consists of phosphatidylinositol, glucosamine, mannoses and phosphoethanolamines. It acts as a lipid anchor for various plasma membrane proteins. GPI-APs play important roles in host self-defense, intercellular signal transduction, and other important processes. In addition, some GPI-APs function as receptors for certain viruses and toxins. The GPI-anchor is widely distributed and conserved in various eukaryotes, and is essential for the development of higher animals, as well as for the growth of yeasts and protozoan parasites. The modification of proteins due to the attachment of the GPI-anchor functions as a protein localization and sorting signal. Our current project is to identify and clarify the functions of all the genes involved in the biosynthesis of the GPI-anchor in the endoplasmic reticulum (ER) (PIG genes; PhosphatidylInositol Glycan), and in the sorting and localization of GPI-APs after their anchorage with GPI (PGAP genes; Post GPI-Attachment to Proteins). We expect that these studies will reveal why many proteins are modified by the GPI-anchor.
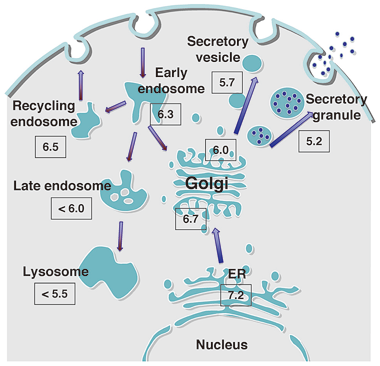 |
Fig. 3 pH regulation of intracellular organelles |
In collaboration with our colleagues in England, we identified a disease called inherited GPI deficiency (IGD) that is caused by the PIGM mutation. PIGM is a mannosyltransferase-encoding gene that plays an essential role in GPI biosynthesis. The recent analysis of the exomes of patients with IGD with the next generation sequencer has already revealed twelve kinds of IGD to date. To further analyze IGDs, we are collaborating with many clinicians and scientists in Japan and abroad. As complete GPI deficiency is embryonically lethal, patients with IGD due to PIG gene mutations only have partial GPI deficiencies. The patients with IGD that is due to remodeling defects even have null mutations. The symptoms of IGD vary depending on the defective genes and the degree of defectiveness. The main symptoms are intellectual disability and seizures that are often accompanied by hyperphosphatasia, abnormal facial features, brachytelephalangy and sometimes other organ anomalies. IGD has been proven to be the main cause of Mabry syndrome/hyperphosphatasia mental retardation syndrome and CHIME syndrome. IGD has also been found in Early Onset Epileptic Encephalopathies such as Ohtahara syndrome and West syndrome. Good diagnostic markers for IGD are the presence of hyperphosphatasia and the decreased flow cytometric expression of granulocyte CD16, which is a GPI-AP . We will establish the diagnostic criteria by accumulating patient information and seek effective therapies.Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired GPI deficiency that is caused by somatic mutation of the X-linked PIGA gene in hematopoietic stem cells. However, it was recently reported that PNH can also be caused by somatic mutation of one allele of the PIGT gene in combination with a germline mutation in the other allele. This showed that PIGA is not always the responsible gene in PNH, although this situation is rarely seen. PNH is a hematopoietic disease that is characterized by the expansion of clonal cells that are defective in GPI biosynthesis. As a result, abnormal erythrocytes lacking the GPI-APs CD59 and DAF/CD55 (which inhibit the activation of complement) predominate, leading to massive hemolysis due to excessive complement attack during infections and other events. We are presently analyzing the pathogenesis of PNH by seeking the mechanism behind the clonal expansion of GPI-deficient hematopoietic stem cells.
Each intracellular organelle is compartmentalized by lipid bilayers and possesses a characteristic environment, proteins and lipids that allow it to fully exercise its functions. A major environment factor is pH. The lumens of organelles in the secretory and endocytosis pathways are known to have acidic environments. This lumen acidity is believed to play critical roles in many biological processes in cells, including the transport, processing, and glycosylation of proteins and lipids. It is also known to influence the morphology of organelles and to participate in the onset and pathology of diseases that are characterized by pH regulation defects. However, the mechanisms behind the latter abnormal phenotypes are largely not known. Recently, we reported for the first time the establishment of mutant cells in which the Golgi pH is dysregulated. Analysis of these cells led to the identification of the gene that bore the mutations and was responsible for the deranged functions of the mutant cells. This project aims to clarify the mechanisms and biological significance of acidic pH regulation by establishing and analyzing additional mutant cells that are defective in organelle pH regulation and to improve our understanding about how such mutations can lead to pathology, as this will facilitate the identification of effective drugs. As an offshoot from the above study, we are also analyzing the components and functions of the membrane contact sites between closely apposed organelle membranes where lipids and ions are transported.
Major publications
- Seong J, Wang Y, Kinoshita T, Maeda Y. Implication of lipid moiety in oligomerization and immunoreactivities of GPI-anchored proteins. J Lipid Res. 2013 Apr;54(4):1077-91
- Murakami Y, Kanzawa N, Saito K, Krawitz PM, Mundlos S, Robinson PN, Karadimitris A, Maeda Y, Kinoshita T. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem. 2012 Feb 24;287(9):6318-25.
- Fujita M, Watanabe R, Jaensch N, Romanova-Michaelides M, Satoh T, Kato M, Riezman H, Yamaguchi Y, Maeda Y, Kinoshita T. Sorting of GPI-anchored proteins into ER exit sites by p24 proteins is dependent on remodeled GPI. J Cell Biol. 2011 Jul 11;194(1):61-75.
- Kanzawa N, Maeda Y, Ogiso H, Murakami Y, Taguchi R, Kinoshita T. Peroxisome dependency of alkyl-containing GPI-anchor biosynthesis in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2009 Oct 20;106(42):17711-6.
- Fujita M, Maeda Y, Ra M, Yamaguchi Y, Taguchi R, Kinoshita T. GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell. 2009 Oct 16;139(2):352-65.